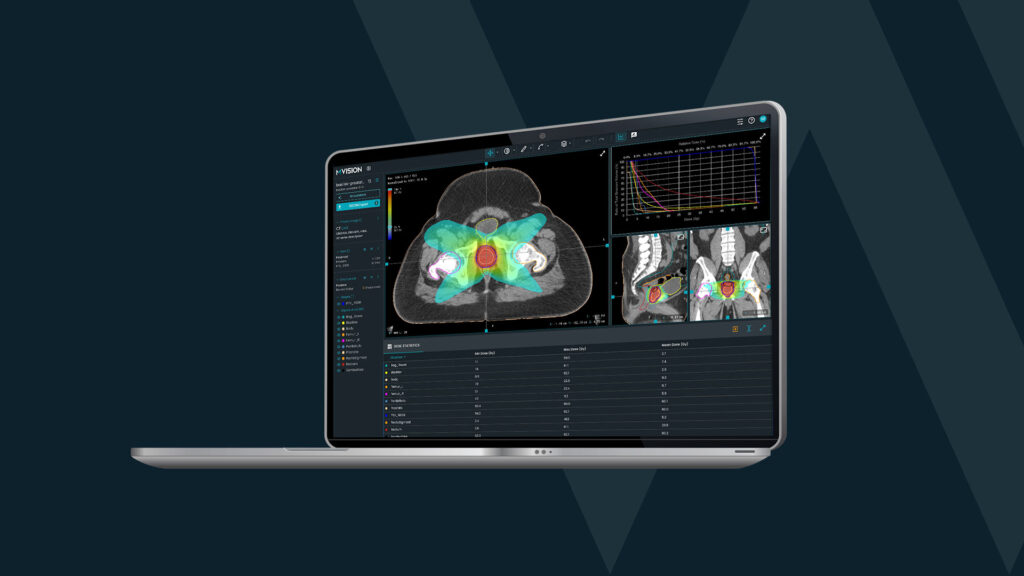
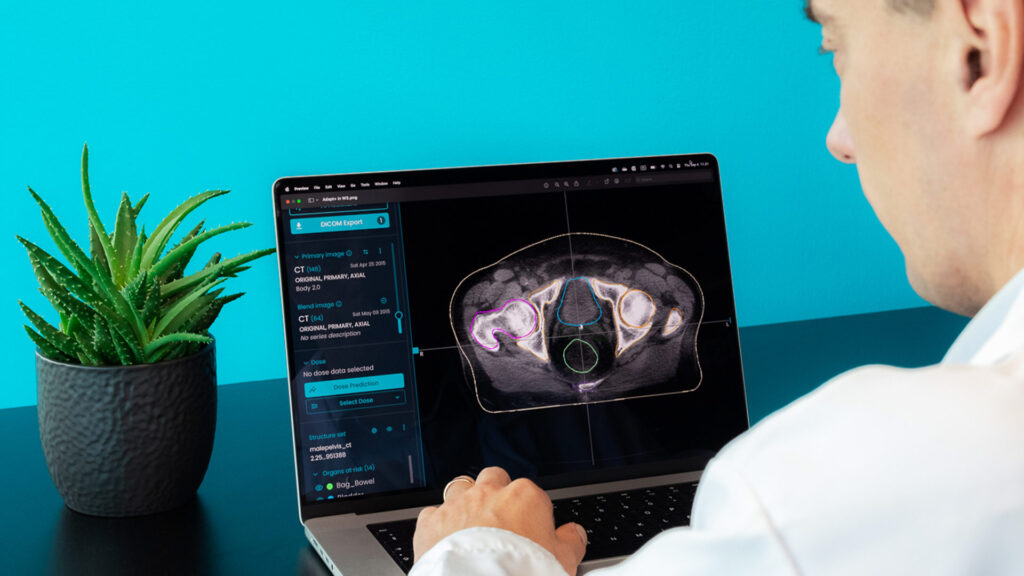
Technology is evolving, and the same applies to Radiation Oncology. New ways and new tools require new rules. Artificial intelligence (AI) brings tremendous potential, so the world has to learn how to get the best of it, safely.
Al systems identified as high-risk have to comply with strict requirements, including risk-mitigation systems, high quality of data sets, logging of activity, detailed documentation, clear user information, human oversight, and a high level of robustness, accuracy, and cyber-security.
MVision offers solutions that are compliant with international regulations, so the Radiation Oncology professionals can use them for the benefit of cancer patients.
Smart solutions
The right to benefit from adequate healthcare is a basic human right, and societies invest considerable effort to ensure that for their citizens. However, the burden of cancer is expected to rise, and the resources are limited, including the workforce. Applying artificial intelligence-based solutions can help to improve accessibility and efficiency of various healthcare interventions.
However, development of new solutions requires new regulations that provide a legal framework aimed to ensure responsible handling of sensitive medical data. Regulations are also establishing common guidelines and standards that are needed to improve compatibility and interoperability. When AI systems adhere to international regulations, their reliability and trustworthiness increase.
Creating a safe frame
According to a recent review of Global regulatory frameworks for the use of Artificial Intelligence in the healthcare services sector, AI in healthcare is currently supervised under the framework of Software as a Medical Device (SaMD). It has to be mentioned that these regulations do not apply to certain AI applications, such as software that provide clinical support or recommendations to healthcare professionals. The reason for this is that individuals are expected to critically analyze the recommendations of the AI application and make their own decisions.
The review focused on regulatory frameworks, legislations, laws, policies, and guidelines and identified two types of laws: hard and soft. The hard laws refer to rules that are legally binding, and the soft laws represent professional guidelines, voluntary standards, codes of conduct, recommendations, agreements, national action plans, or policy documents. Most of the regulations identified by the review can be included in the soft-law category. For developing and enforcing hard-laws, the national-level official entities are responsible, such as governments. For the soft-laws, the relevant stakeholders are the developers and the users. “Developers” are persons or organizations involved in the planning, funding, developing, and/or maintaining AI-medical devices, and “users” refers to a singular person or an organization that uses AI-medical devices in the delivery of healthcare services.
The soft-laws can be easily amended given the evolving landscape of AI technologies. However, since these are voluntary guidelines, the organizations have the option to not adopt them.
European AI Act
The European Union (EU) regulations on AI use in healthcare had a roadmap that started in 2019, with the “Ethics Guidelines for Trustworthy AI” and the “Policy and Investment Recommendations”. The “European Medical Device Regulation”, wherein the risk classification of SaMDs was based on diagnostic and therapeutic intentions, was published in 2021.
The EU AI Act, a harmonized legal framework for AI products and services from the development phase to their application, was proposed in the same year and came into force in 2024. Similarly to other international regulations, the AI Act uses a risk-based approach. In high-risk AI systems used in the healthcare sector, the manufacturer has to ensure data governance and risk management.
High-risk AI systems have to comply with the following requirements:
- Data Governance: High-risk AI systems must be trained on high-quality datasets that are relevant, representative, and free from bias to prevent discriminatory outcomes.
- Documentation and Traceability: Developers must maintain detailed technical documentation to ensure the AI system’s compliance and allow for thorough audits.
- Transparency: Users must be informed about the AI system’s capabilities, limitations, and the potential risks. Systems must be explainable, enabling users to understand and challenge decisions made by AI.
- Human Oversight: AI systems must allow human intervention to minimize risks and prevent harmful outcomes. For instance, decisions made by AI should be overseen by humans who can override the system if necessary.
- Robustness, Accuracy, and Security: AI systems must be resilient and able to function correctly even under adverse conditions. They should be designed to minimize errors and security risks.
Important dates:
- 1st of August 2024 – The EU AI Act came into force.
- 2nd of August 2025 – Deadline for the Member States to designate national competent authorities, who will oversee the application of the rules for AI systems and carry out market surveillance activities.
- 2nd of August 2026 – Date on which the majority of rules of the AI Act will start applying. However, prohibitions of AI systems deemed to present an unacceptable risk will already apply after six months, while the rules for so-called general-purpose AI models will apply after 12 months.
Companies not complying with the rules will be fined up to 7% of the global annual turnover for violations of banned AI applications, up to 3% for violations of other obligations and up to 1.5% for supplying incorrect information.
The Commission has launched the AI pact, aimed to bridge the transitional period before full implementation. Through the AI pact, AI developers are invited to voluntarily adopt key obligations of the AI Act ahead of the legal deadlines.
Key players for the AI act application:
- The Commission’s AI Office – the key implementation body for the AI Act at EU-level, as well as the enforcer for the rules for general-purpose AI models.
- The European Artificial Intelligence Board – ensures a uniform application of the AI Act across EU Member States and will act as the main body for cooperation between the Commission and the Member States.
- Scientific panel of independent experts – will offer technical advice and input on enforcement. In particular, this panel can issue alerts to the AI Office about risks associated with general-purpose AI models.
- Advisory forum – composed of a diverse set of stakeholders.
The international landscape
According to the above-mentioned review, pioneers in the area of regulating AI in healthcare are Europe, the United Kingdom (UK), the United States of America (USA), Australia, China, Brazil and Singapore.
In the USA, the FDA evaluates AI-based technologies under the existing regulatory framework for medical devices. The “AI/ML-based SaMD Action Plan” was issued in January 2021, outlining five actions based on the total product life cycle approach:
- Specific regulatory framework
- Good machine learning practices;
- Patient-centric approach, including the transparency of devices to users;
- Methods for the elimination of ML algorithm bias and algorithm improvement;
- Real-world performance monitoring pilots.
Good machine learning practices refer to:
- High relevance of available data to the clinical problem and current clinical practice;
- Consistency in data collection that does not deviate from the SaMD’s intended use;
- Planned modification pathway;
- Appropriate boundaries in the datasets used for training, tuning, and testing the AI algorithms;
- Transparency of the AI algorithms and their output for users
In the UK, the “Evidence Standards Framework for Digital Health Technologies” was published in 2019, as a result of the collaboration of the National Institution for Health and Care Excellence (NICE) and the National Health Service (NHS) England. It provides the regulations for software, apps and online platforms that can be combined with other health products or used as standalone products.
The Medicines and Healthcare Products Regulatory Agency (MHRA) established a regulatory reform programme known as the “Software and AI as a Medical Device Change Programme” in September 2021. The topics included cybersecurity and data privacy risks, a post-market evaluation of the medical device and additional challenges that AI can pose, including evolving AI algorithms, bias, and interpretability.
In Australia, “Regulatory changes for software-based medical devices” was published in August 2021, to explain the amendments to the Therapeutic Goods (Medical Devices) Regulation including a risk-based classification approach, and updated In May 2024.
In Brazil, the draft AI law issued in December 2022 classifies health applications as high-risk AI systems. The providers need to maintain a publicly accessible database that provides the details of the completed risk assessments and conduct periodically repeated algorithmic impact assessments. It also establishes governance structures that facilitate the individuals’ rights to information, explanation, challenge, human intervention, non-discrimination, the correction of discriminatory bias, privacy, and the protection of personal data. The providers are strictly liable for any damages caused by their AI system, according to the same document.
In China, the Centre for Medical Device Evaluation under the NMPA published the “Guidelines for Registration and Review of Artificial Intelligence-Based Medical Devices” on 7 March 2022. The aim was to standardize the quality management of software and cybersecurity of medical devices at the national level, but it is also harmonizing at the international level.
In Singapore, Health Sciences Authority (HSA) released a second revision of its “Regulatory Guidelines for SaMD—A Lifecycle Approach” in April 2022. It highlights that the developers need to provide intended purpose, input data details, specifications of performance, control measures, and post-market monitoring and reporting. The “AI in Healthcare Guidelines” published in October 2021 provides recommendations on good practices for AI developers and AI implementers, based on the principles adapted from the AI Governance Framework established by the Personal Data Protection Commission (PDPC).
MVision’s compliance with international requirements
Compliance of MVision AI Contour+
- Patient-centric approach, including the transparency of devices to users;.
- Transparency implies being open about the process details that are relevant for the users. MVision closely follows the international contouring guidelines for developing Contour+. Having this approach, the users can rely on what has been used as reference. MVision also includes relevant information about the algorithm limitations in the instructions for use, and shares more details about the development and test data annotation processes in the on-boarding sessions for the customers.
- Methods for the elimination of ML algorithm bias and algorithm improvement;
- Using a non-representative sample of data can induce bias. MVision carefully chooses the training and test data samples, to cover various situations found in the clinical practice. Moreover, the initial product performance is tested in diverse settings. Potential influence of certain outlier elements is evaluated and if needed, corrections being made, by adding other datasets.
- Real-world performance monitoring pilots.
- MVision team continuously receives users’ feedback and encourages independent clinical validation studies, learning from their results. Apart from short-term projects, MVision is participating in a 3-year UK NICE health technology evaluation to reveal the real-world benefits in this evidence creation program.
Good machine learning practices refer to:
- High relevance of available data to the clinical problem and current clinical practice;
- MVision uses development and test data from more than 40 countries, including all major scanner manufacturers and models. Datasets are coming from patients having a wide age range (from below 10 up to 90 years), yielding anatomical variations and image artifacts.
- Consistency in data collection that does not deviate from the SaMD’s intended use;
- Data collection is consistent with the intended use, which is the auto-segmentation purpose for radiation therapy.
- Planned modification pathway;
- MVision has planned modifications for minor and major changes. Contour+ alone has been updated ten times, always with a clear change plan before modification
- Appropriate boundaries in the datasets used for training, tuning, and testing the AI algorithms;
- The dataset splits are created at the patient level, i.e. all data from a patient can be only in a single data split (e.g. training, validation, testing, etc.). The details of the data splits are provided in the data management plan document.
Performance validations were performed by several qualified radiation therapy experts in diverse geographical locations to ensure our product consistently performs in clinically relevant conditions. Their results have been published in peer-reviewed journals or presented in international conferences.
References
- Palaniappan K, Lin EYT, Vogel S. Global Regulatory Frameworks for the Use of Artificial Intelligence (AI) in the Healthcare Services Sector. Healthcare (Basel). 2024 Feb 28;12(5):562. doi: 10.3390/healthcare12050562. PMID: 38470673; PMCID: PMC10930608.
- European Artificial Intelligence Act comes into force. European Commission Press Release. Accessed on 28 August 2024. Available at: https://ec.europa.eu/commission/presscorner/detail/en/ip_24_4123
- US Food & Drug Administration. Good Machine Learning Practice for Medical Device Development: Guiding Principle. Accessed on 16 October 2024. Available at: https://www.fda.gov/media/153486/download
- Australian Government. Artificial Intelligence (AI) and medical device software. Information for software manufacturers about how we regulate AI medical devices. Accessed on 16 October 2024. Available at: https://www.tga.gov.au/how-we-regulate/manufacturing/manufacture-medical-device/manufacture-specific-types-medical-devices/artificial-intelligence-ai-and-medical-device-software